HIV-1
PROJECTS
The acquired immunodeficiency syndrome (AIDS) continues to spread unchecked since its first documentation in 1981. Twenty-five years later, nearly 50 million individuals are living with HIV/AIDS world wide, according to the figures released by the Joint United Nations Program on HIV/AIDS (UNAIDS) and World Health Organization (WHO).
Given the importance and extent of the HIV epidemic around the world, studies
to achieve a better understanding of underlying viral pathways that prevent us
from finding a cure for this disease are of crucial importance. Extensive
research and clinical trials have led to the discovery that certain enzymes
such as protease (PR), reverse transcriptase (RT) and integrase (IN), are of crucial importance
for viral infectivity. HIV protease is an aspartic protease involved
in the processing of the viral Gag and Gag/Pol polyproteins; this step
is essential for gaining viral infectivity. Without an active protease
the virion is unable to maturate and infect other CD4+ cells. Currently, there are eight
protease inhibitors (PIs) used to treat HIV-positive individuals. These PIs are
administered in conjunction with a combination of RT inhibitors and/or integrase inhibitors
and/or fusion inhibitors, and this cocktail, called HAART, has improved the
prognosis of HIV-infected patients.
But, even with this complex
therapeutic approach, the development of drug resistance poses the greatest
challenge for treating the HIV infection and the margin of success for
achieving and maintaining virus suppression is narrow. At the
molecular level, resistance to PIs predominantly takes the form of mutations
within the PR molecule that preferentially lower the affinity of PIs with
respect to protease substrates, while still maintaining viable catalytic
activity. Basically,
resistant mutations arise because they provide the virus with an advantage to
survive in the presence of the drug. A number of resistance mutations have been
characterized for all clinically used inhibitors, including atazanavir, the
most recent one on the market. These difficulties only add to the significant
genetic diversity that characterizes HIV. While HIV-1 subtype B has been the
most widely studied, subtypes A, C, and D predominate worldwide. Among the approximately 40 million people living with HIV/AIDS in 2003, more
than 80% were infected with HIV-1 non-B subtypes. Substitutions at the positions designed as secondary
resistance mutations in subtype B occur at high rates in certain non-subtype B
viruses as naturally occurring or baseline polymorphisms.
Whereas in treatment-naïve patients many of these baseline polymorphisms do
not confer resistance to drugs per se among different clades, they may
facilitate the development of drug resistance. Recent studies have shown that
non-B isolates were statistically associated with more rapid progression to
resistance after antiretroviral therapy and they had different
mutational patterns as compared to B isolates. The combined effect of naturally
existing polymorphisms and drug resistant mutations might have important
consequences on the feasibility of continuing to use current HIV-1 protease
inhibitors for non-subtype B infections.
Our HIV protease-related research develops into two main directions: (1)
analysis of primary and secondary drug resistance mutations within HIV
protease and (2) phylogenetic diversity of HIV-1 protease and its impact
on response to current anti HIV therapy. Our lab has performed extensive biochemical
and structural studies to identify and characterize primary and secondary
mutations.
A
student working on this project will learn how to:
- express the HIV protease in E. coli expression system, to
extract, refold to the native conformation, and purify the enzyme.
- measure enzyme kinetic properties (Km, kcat, kcat/Km) and inhibition (Ki) with all clinically used inhibitors and for other inhibitors undergoing clinical trials. The student will also learn how to assay enzymatic activity using a chromogenic substrate that mimics the natural substrate of the enzyme. To further study each different protease, determination of kinetic constants would be employed: Km, kcat, and Ki.
- crystallize and solve the three-dimensional structure of the
different variants of HIV protease, alone or complexed with inhibitors.
Working on this project: Roxana Coman graduate student
Taylor Gilliland undergraduate student
Marty Fernandez undergraduate student
Kajia Zhu undergraduate student
2. CHARACTERIZATION OF THE PROPERTIES OF THE GAG-POL PROTEINS OF NON-B SUBTYPES. Processing of these precursor polyproteins (Figure 1) is accomplished by the viral PR encoded within Gag/Pol without assistance from cellular proteases and occurs at the cell membrane during virion packing. Both the order and kinetics of cleavage as well as the extent of precursor processing appear to be critical steps in the generation of fully infectious, correctly assembled viral particles.
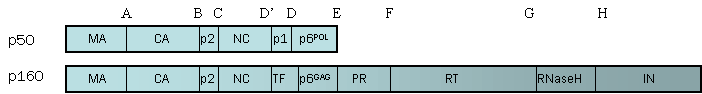
Figure 1. Gag and Gag/Pol Cleavage Sites. The HIV protease cleaves 5 sites within Gag, 11 sites within Gag/Pol polyproteins, and 1 cleavage site within accessory protein Nef (not shown).
We focus on the influence of
mutations within the Gag/Pol fusion protein on the efficiency and order of PR
processing. Our current work has identified several points within the region
from the start of protein p2 through the end of the p6Pol where
mutations affect processing. We will employ this
in vitro transcription/translation
system to screen natural variants for altered processing patterns. We will
pursue the analysis of the effects of antiviral inhibitors on the processing
pathway and rate. This will begin with the clinically approved compounds now
available from the NIH AIDS Research
& Reference Reagent Program,
but will also include new compounds that we develop or discover in our studies.
All these goals will be accomplished in collaboration with Dr. Maureen
Goodenow.
For a student to be able to work on this project, he/she must take the radioactivity safety class offered by UF.
Working on this project: Roxana
Coman graduate student
3. EXPLORATION OF THE PROPERTIES AND THE STRUCTURE OF A VARIETY OF EXTENDED FORMS OF HIV-1 PROTEASE. These are constructs containing the protease itself and increasingly longer sequences upstream of the protease. Our studies focus on the N-terminal end of the Gag/Pol protein sequence, with protease located at the C-terminus of the constructs, as we know that the initial steps in processing occur in the region upstream of protease. We hypothesize that there is a structural pre-organization of Gag/Pol that brings the C cleavage site [p2/NC] near the active site cleft of the protease, resulting in the initial rapid cleavage of that junction (Figure 2). If this structure can be observed by crystallography, through the construction of an inactive protease-Gag fusion protein, then we will identify a new target for drug discovery, i.e., the interaction surface for formation of the pre-cleavage structure.
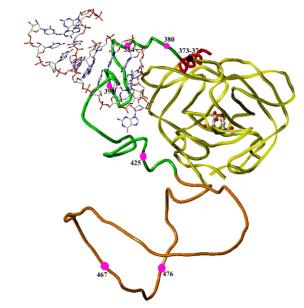
Figure 2. Model of the region from p2 (red) through protease (yellow), with NC (green) and transframe-p6Pol intervening (orange). MA-CA is not shown in this model. This model was built using the structures of the protease (yellow tube, with catalytic aspartic residues in ball-and-stick representation, and the structure of NC (green tube) bound to DNA (shown in ball-and-stick representation) [1]. The p2 segment is represented as a helix (red) and the TR-p6Pol is shown as a coil (orange). This structure brings the p2/NC cleavage junction near the active site cleft of the protease and would be a possible intermediate just before the start of Gag/Pol self-processing. The second p2/NC/TF segment could be disordered at this point, awaiting successful cleavage of the first segment, and is not shown in this figure. Red balls in the figure show points where mutations have been found to alter Gag-Pol processing. Two points in the p2 segment are represented by a black ball.
We will try to express, purify, refold and crystallize two variants: the active
p2-PR construct and the inactive D25N variants.
Working on this project: Lashara Livingston undergraduate student
Chriss Lazo undergraduate student